Article Type: Case Report, Volume 3 Issue 1
*Corresponding author: Senay Topsakal
Department of Endocrinology and Metabolism, Faculty of Medicine, Pamukkale University, Denizli, Türkiye.
Email: stopsakal@pau.edu.tr
Received: Mar 03, 2026 Accepted: Mar 26, 2026 Published: Apr 02, 2026
Citation: Topsakal S, Taban S, Celik I. Aggressive pituitary macroadenoma causing refractory Cushing’s disease: A 14-year follow-up. Ann Case Rep Med Images. 2026; 3(1): 1077.
Copyright: Topsakal S et al. © All rights are reserved
Cushing’s Disease (CD) is most commonly caused by ACTH-secreting pituitary microadenomas, whereas macroadenomas are rare and often associated with aggressive behavior, treatment resistance, and poor prognosis. We report a challenging case of CD caused by a pituitary macroadenoma with an unfavorable long-term outcome. A 38-year-old woman presented with Cushingoid features and metabolic comorbidities. Biochemical evaluation confirmed CD, and pituitary MRI revealed a macroadenoma without initial visual field defects. Despite multimodal treatment—including transsphenoidal surgery, Gamma Knife radiosurgery, repeat neurosurgical interventions, and medical therapies—biochemical remission was never achieved. The disease course was marked by progressive tumor recurrence, severe metabolic complications, and neurological events over a 14-year follow-up period. The patient ultimately died following a cerebrovascular event. This case highlights the aggressive nature and therapeutic challenges of ACTH-secreting pituitary macroadenomas. Durable remission may not be achievable despite intensive multimodal therapy, underscoring the need for early diagnosis, individualized management, and vigilant long-term follow-up in refractory CD.
Keywords: Cushing’s disease; Pituitary macroadenoma; Long-term outcome.
Cushing’s Disease (CD), the most common cause of endogenous Cushing’s Syndrome (CS), results from an Adrenocorticotropic Hormone (ACTH)-secreting pituitary adenoma, leading to chronic hypercortisolism. While approximately 80% of these tumors are microadenomas (<10 mm), a smaller proportion—10% to 20%—are macroadenomas (≥10 mm), which are often associated with more aggressive clinical behavior, higher tumor burden, and increased rates of treatment resistance and recurrence [1-3].
Patients with pituitary macroadenomas frequently present with more pronounced symptoms and complications related to both hypercortisolism and mass effect. These tumors also pose significant diagnostic and therapeutic challenges, often requiring multimodal interventions including surgery, radiotherapy, and medical therapy. Despite these efforts, achieving sustained biochemical remission remains difficult in a subset of patients [4-9].
In this report, we present a case of Cushing’s disease due to a pituitary macroadenoma in a female patient who was followed for over a decade in our center. This patient represents the longest follow-up period among macroadenoma-related CD cases in our clinic. Her clinical course was particularly complex due to persistent hypercortisolism, multiple surgical and medical treatment failures, and severe metabolic complications. The aim of this case report is to present a patient with Cushing’s disease due to a pituitary macroadenoma, who had the longest duration of follow-up in our clinic and whose management was particularly challenging. By sharing this complex clinical course, we aim to underscore the heterogeneity of macroadenoma-related Cushing’s disease, the limitations of existing therapeutic modalities, and the critical role of long-term, multidisciplinary management in refractory cases.
This case report presents a 38-year-old female patient who was admitted to our outpatient clinic in 2006 with complaints of fatigue, weakness, and persistent headache. Her medical history included type 2 diabetes mellitus, hypertension, and nodular goiter. On physical examination, she exhibited a moon face and central obesity, indicative of a Cushingoid appearance. The patient was diagnosed with Cushing’s disease based on clinical features, biochemical hypercortisolism, and pituitary MRI findings consistent with a macroadenoma. Initial imaging revealed a sellar mass invading the right cavernous sinus without visual field impairment. Despite transsphenoidal surgery followed by Gamma Knife radiosurgery, biochemical remission was not achieved.
During long-term follow-up, serial MRI examinations demonstrated residual and recurrent tumor growth with persistent cavernous sinus invasion. Repeated hormonal assessments showed sustained ACTH-dependent hypercortisolism, with progressively increasing urinary free cortisol levels. Dynamic testing failed to demonstrate adequate cortisol suppression.
Medical therapies, including ketoconazole and pasireotide, were limited by poor tolerability and insufficient biochemical response. The disease course was complicated by worsening insulin-resistant diabetes, uncontrolled hypertension, and recurrent neurological events. Additional surgical interventions, including repeat transsphenoidal and transcranial approaches, failed to achieve disease control.
Over a 14-year follow-up period, persistent hypercortisolism and progressive tumor behavior led to severe metabolic and vascular complications. The patient ultimately died following a cerebrovascular event. Key radiological, hormonal, metabolic, and therapeutic features of the disease course are summarized in (Figure 1, Tables 1 and 2).
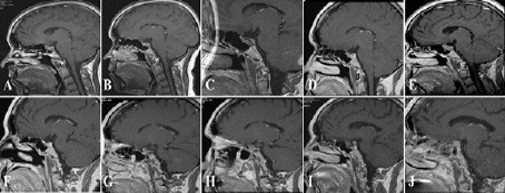
Figure 1: Representative sagittal post-contrast T1-weighted pituitary MRI images demonstrating the longitudinal evolution of the pituitary macroadenoma over a 16-year follow-up period. Images were obtained in (A) 2006, (B) 2007, (C) 2008, (D) 2009, (E) 2012, (F) 2013, (G) 2014, (H) 2015, (I) 2018, and (J) 2022, illustrating residual disease, progression, and recurrence despite multimodal therapy.
| Year | Imaging findings | Tumor characteristics | Intervention / Clinical event |
|---|---|---|---|
| 2005 | Initial pituitary MRI | 1.5×2 cm macroadenoma | First transsphenoidal surgery |
| 2006 | Postoperative MRI | Residual tumor, cavernous sinus invasion, ICA encasement | Gamma Knife radiosurgery |
| 2007-2011 | Serial MRIs | Partial cystic regression | — |
| 2012 | MRI | Progressive macroadenoma (18×16×20 mm) | — |
| 2013 | MRI | Persistent invasive tumor | Second Gamma Knife |
| 2015-2016 | MRI stable | Cavernous sinus involvement | Facial paralysis episodes |
| 2018 | MRI | Residual adenoma (13×10 mm) | Repeat transsphenoidal surgery |
| 2019 | MRI | Persistent invasive disease | Transcranial surgery |
| 2020 | — | — | Cerebrovascular event (death) |
| Parameter | Key findings over time |
|---|---|
| Basal ACTH | Persistently elevated, progressive increase |
| Serum Cortisol | Inadequate suppression on LDDST/HDDST |
| Urinary Free Cortisol | Marked increase during follow-up |
| CRH Test | Inadequate cortisol and ACTH response |
| Glycemic Status | Severe insulin resistance, poor control |
| Bone Mineral Density | Mild osteopenia, no severe osteoporosis |
| F-18 FDG PET-CT | Non-specific liver uptake |
| Ga-68 DOTA-TATE PET-CT | No pathological uptake |
Cushing’s Disease (CD) caused by pituitary macroadenomas represents a distinct and more aggressive clinical entity compared with microadenoma-related disease. Tumor size, invasiveness, and underlying molecular alterations critically influence treatment response and long-term outcomes. In particular, cavernous sinus invasion and carotid artery encasement, as observed in our patient, markedly reduce the likelihood of complete surgical resection and durable biochemical remission [2-5].
From a mechanistic perspective, corticotroph macroadenomas differ biologically from microadenomas. While USP8 mutations—frequently detected in microadenomas—enhance EGFR signaling and are associated with smaller tumor size and better responsiveness to medical therapies such as pasireotide, macroadenomas are more often USP8–wild type and may harbor alternative genetic alterations, including TP53 mutations [3,5-7]. TP53-associated tumors exhibit increased proliferative activity, invasiveness, treatment resistance, and poor survival. The aggressive clinical course, repeated recurrences, and lack of response to surgery, radiosurgery, and medical therapy in our case are consistent with this unfavorable molecular profile.
Excessive and sustained cortisol exposure further amplifies disease severity by driving profound metabolic and vascular complications. Chronic hypercortisolism promotes insulin resistance, hypertension, endothelial dysfunction, and a prothrombotic state, substantially increasing cerebrovascular risk [6-13]. Despite intensive multimodal treatment, persistent hypercortisolism in our patient likely contributed to cumulative cardiovascular and neurological damage, ultimately resulting in a fatal cerebrovascular event.
Although transsphenoidal surgery remains first-line therapy, remission rates in macroadenoma-related CD are substantially lower than in microadenomas. Adjuvant radiotherapy and medical treatments are frequently required, yet their efficacy is often limited and delayed [14-18]. This case illustrates that even aggressive multimodal strategies may fail when unfavorable tumor biology predominates.
Overall, this report underscores that pituitary macroadenoma–related CD is driven not only by tumor size but also by intrinsic molecular aggressiveness. Early identification of high-risk biological features and integration of personalized, mechanism-based therapeutic approaches are essential to improve outcomes in this challenging subgroup of patients.
Cushing’s disease caused by pituitary macroadenomas is a particularly aggressive and treatment-resistant entity, characterized by invasive growth, frequent recurrence, and substantial long-term morbidity. This case highlights that conventional therapeutic strategies may fail to achieve durable disease control when unfavorable tumor biology predominates. Early integration of detailed biochemical and molecular characterization is therefore critical to identify high-risk patients and guide personalized treatment decisions. Effective management requires a truly multimodal approach, combining experienced pituitary surgery, appropriately selected adjuvant therapies, and meticulous long-term surveillance. Advances in molecular profiling and targeted therapeutics offer promising opportunities to shift the current treatment paradigm and improve outcomes in this challenging and often devastating form of Cushing’s disease.
Author contribution: Senay Topsakal: Conceptualization, Supervision Data curation, Formal analysis, Writing—original draft, Writing—review & editing. S Taban: Conceptualization, Formal analysis, Writing— review & editing. I Celik: Data curation, Writing—review & editing. All the authors reviewed the manuscript and approved its final version.
Conflict of interest : There is no conflict interest.