Article Type: Case Report, Volume 3 Issue 1
*Corresponding author: İ Merve Uçar Baytaroğlu
Rheumatology Department, Uşak Training and Research Hospital, Uşak, Türkiye.
Email: ucarmerve1988@hotmail.com
Received: Jan 21, 2026 Accepted: Feb 13, 2026 Published: Feb 20, 2026
Citation: Baytaroglu IMU, Cetinkaya AM. Metastatic merkel cell carcinoma in a patient with systemic lupus erythematosus and antiphospholipid syndrome. Ann Case Rep Med Images. 2026; 3(1): 1067.
Copyright: Baytaroglu IMU et al. © All rights are reserved
Introduction: Merkel Cell Carcinoma (MCC) is a rare, aggressive cutaneous neuroendocrine malignancy strongly associated with immunosuppression. We report the third documented case of MCC in a patient with Systemic Lupus Erythematosus (SLE), highlighting the critical role of chronic immunosuppressive therapy rather than the autoimmune disease itself as the primary risk factor for MCC development.
Case presentation: A 52-year-old woman with SLE and Antiphospholipid Syndrome (APS), treated with immunosuppressive agents including azathioprine, rituximab, and corticosteroids for approximately 10 years, presented with a rapidly growing mass in her left lower jaw. Physical examination revealed a firm, fixed, 7 cm mass with overlying skin necrosis. Histopathology confirmed MCC, and PET-CT demonstrated metastatic disease in regional lymph nodes and liver (Stage IV). The patient received palliative radiotherapy and chemotherapy with cisplatin and etoposide. Following chemotherapy, she developed profound pancytopenia. While managed empirically as an SLE-related hematological flare with high-dose corticosteroids and intravenous immunoglobulin, bone marrow metastasis could not be excluded without biopsy. The patient died from multi-organ failure and respiratory compromise due to tumor progression.
Conclusion: Long-term immunosuppressive therapy likely constitutes the primary risk factor for MCC in autoimmune disease patients. This case emphasizes the importance of dermatological surveillance in chronically immunosuppressed patients and highlights the diagnostic challenges in attributing cytopenias in complex oncology patients with underlying autoimmune diseases.
Keywords: Merkel cell carcinoma; Systemic lupus erythematosus; Immunosuppression; Antiphospholipid syndrome; Pancytopenia; Case report.
Merkel Cell Carcinoma (MCC) is a rare and aggressive cutaneous neuroendocrine malignancy with an incidence of approximately 0.7 per 100,000 person-years. Established risk factors include advanced age, cumulative ultraviolet exposure, fair skin, male sex, and importantly, immunosuppression [1-3]. Approximately 60-80% of MCC cases harbor Merkel Cell Polyomavirus (MCPyV), which drives oncogenesis through viral T antigen expression. Intact T-cell immunity is critical for controlling MCPyV-positive tumors, as viral oncoproteins are highly immunogenic foreign antigens that should trigger robust immune surveillance [2,4].
The link between immunosuppression and MCC is wellknown. Solid organ transplant recipients have a 23.8-fold higher risk of MCC, mainly due to immunosuppressive drugs, while HIV/AIDS patients experience a 13-fold increase [3,5]. Importantly, among autoimmune disease patients, the pattern suggests that treatment intensity, rather than the disease itself, drives MCC risk. Rheumatoid arthritis alone shows a non-significant Standardized Incidence Ratio (SIR) of 2.42 (95% CI: 0.96-5.01), whereas ankylosing spondylitis, which often requires more aggressive immunosuppression, has a significantly higher SIR of 15.62 (95% CI: 2.95-46.25) [5]. TNF inhibitors have been specifically linked to MCC development in autoimmune disease patients [6].
Systemic Lupus Erythematosus (SLE) is a chronic autoimmune disease characterized by immunological dysregulation and multi-organ involvement. Antiphospholipid Syndrome (APS) is an acquired prothrombotic state associated with thrombosis and pregnancy losses. While patients with SLE have an increased overall malignancy risk, this appears largely attributable to the immunosuppressive therapies used to manage the disease rather than to SLE itself. To date, only two cases of MCC in SLE patients have been reported in the medical literature, both with aggressive courses and fatal outcomes [7,8]. We present a third case of MCC arising in a patient with SLE and APS, with particular focus on the role of chronic immunosuppression and the diagnostic challenges in managing treatmentrelated cytopenias.
A 52-year-old female patient was diagnosed with SLE and APS in 2013. Her SLE manifestations included arthritis, arthralgia, and hematological involvement consisting of pancytopenia and refractory thrombocytopenia. Her APS was characterized by five pregnancy losses. Laboratory findings at diagnosis showed positive ANA, persistently low C3 and C4 complement levels, and positive anti-ENA antibodies for centromere, PM100, and Ku antigens. Comorbidities included hypertension. She had no history of smoking or alcohol use. Family history was notable for her father having an intracranial glial tumor. There was no history of excessive sun exposure or prior skin malignancies.
Over the course of approximately 10 years (2013-2023), the patient received multiple immunosuppressive agents for SLE management: hydroxychloroquine (200 mg twice daily, ongoing), azathioprine (100-150 mg daily for approximately 5 years, discontinued 2022), rituximab (two cycles of 1000 mg infusions, 2018 and 2020), and Intravenous Immunoglobulin (IVIG, multiple courses for refractory thrombocytopenia). She frequently required corticosteroid escalation during attempted tapers due to disease flares, resulting in prolonged periods of prednisone exposure averaging 10-20 mg daily with intermittent high-dose pulses.
In May 2025, the patient noted a rapidly growing mass in her left lower jaw area. On physical examination, a firm, fixed, non-tender mass measuring approximately 7 cm in greatest dimension was palpable in the left submandibular region extending toward the mandibular angle. The overlying skin showed erythema with focal areas of necrosis and ulceration (Figure 1). There was no facial nerve weakness. Palpable lymphadenopathy was present in the left cervical chain (levels I-II). The remainder of the examination was unremarkable; there were no other suspicious skin lesions, and no hepatosplenomegaly was appreciated.
Timeline
2013: Diagnosis of SLE and APS; initiation of hydroxychloroquine and corticosteroids
2013-2017: Addition of azathioprine for steroid-sparing; recurrent flares requiring steroid escalation
2018: First rituximab cycle for refractory thrombocytopenia
2020: Second rituximab cycle; multiple IVIG courses
2022: Azathioprine discontinued due to persistent cytopenias
May 2025: Patient noticed rapidly growing left jaw mass June 2025:MRI showed 55×33 mm subcutaneous mass; biopsy performed
August 2025: Histopathology confirmed Merkel cell carcinoma
September 2025: PET-CT staging revealed metastatic disease (Stage IV); palliative radiotherapy initiated
October 2025: Chemotherapy started; profound pancytopenia developed after first cycle
November 2025: Progressive deterioration; death from multi-organ failure and respiratory compromise
Diagnostic assessment
A neck/face MRI performed in June 2024 revealed a mass lesion measuring 55×33 mm, confined to the subcutaneous layer adjacent to the left mandibular ramus, with no bone invasion. Incisional biopsy was performed, and histopathologic examination in August confirmed the diagnosis of Merkel cell carcinoma. Immunohistochemistry demonstrated positivity for CK20 (perinuclear dot pattern), synaptophysin, and chromogranin A, with negativity for CK7 and TTF-1, consistent with primary cutaneous MCC. MCPyV testing was not performed.
Staging PET-CT in September demonstrated hypermetabolic activity (SUVmax: 20.1) in an irregularly contoured mass approximately 69×64×95 mm located inferior to the left mandible, representing the primary malignancy. Hypermetabolic activity (SUVmax: 13.5) was observed in metastatic lymph nodes in the left submandibular region (largest node: 18 mm short axis). Hepatic metastases were also identified. No bone metastases were evident on PET-CT. Based on the AJCC 8th edition staging system, the patient was classified as Stage IV (T4N1bM1) MCC.
At the time of oncological evaluation, laboratory investigations revealed: platelet count 20,000/μL (normal: 150,000400,000), neutrophil count 2,690/μL (normal: 1,500-8,000), hemoglobin 11.5 g/dL (normal: 12-16), erythrocyte sedimentation rate 27 mm/hour, low C3 (0.52 g/L; normal: 0.9-1.8) and C4 (0.08 g/L; normal: 0.1-0.4) complement levels, negative antidsDNA antibodies, and negative lupus anticoagulant. Antiphospholipid antibodies remained positive: anticardiolipin IgG 51.5 U/mL (normal: 0-10) and anti-β2-glycoprotein-1 IgG 70.1 U/ mL (normal: 0-5). Direct Coombs test was negative. Peripheral blood smear showed thrombocytopenia without schistocytes or leukoerythroblastic changes.
The pancytopenia observed in this patient posed a diagnostic challenge with three main considerations. First, chemotherapy-induced myelosuppression, which is expected with platinum-based regimens; second, bone marrow metastasis from MCC, documented in immunosuppressed patients including the two previously reported MCC-SLE cases; and finally, SLE-related hematological flare, given the patient’s history. A bone marrow biopsy would have been valuable for a definitive diagnosis but was not performed due to the patient’s clinical instability and thrombocytopenia.
Following multidisciplinary tumor board discussion, azathioprine was discontinued. Checkpoint inhibitor immunotherapy (pembrolizumab), the standard first-line treatment for advanced MCC, was considered but deferred due to concerns about triggering SLE flare in the context of baseline hematological abnormalities and low complement levels.
Palliative Radiotherapy (pRT) was initiated targeting the primary tumor (30 Gy in 10 fractions over 2 weeks). Zoledronate (4 mg IV monthly) was started for bone protection. After moderate improvement in blood counts following pRT (platelet count increased to 45,000/μL), the patient was started on chemotherapy with cisplatin (25 mg/m² IV) and etoposide (50 mg/m² IV) weekly for 3 planned cycles.
Following the first chemotherapy cycle, the patient developed profound pancytopenia with a platelet count nadir of 4,000/μL, hemoglobin 8.2 g/dL, and neutrophil count 800/μL. Given the clinical context and the patient’s history, the pancytopenia was empirically treated as a possible SLE-related hematological flare. Corticosteroid therapy was initiated at prednisone 1 mg/kg/day (60 mg daily). Due to inadequate response after 48 hours, intravenous methylprednisolone pulse therapy (500 mg daily for 3 days) and IVIG (1 g/kg over 2 days) were added.
By the fifth day of immunosuppressive therapy, the platelet count increased from 4,000/μL to 17,000/μL. The patient did not experience clinical bleeding during the severe thrombocytopenia period. Radiotherapy was tolerated without significant acute toxicity. The single cycle of chemotherapy given was complicated by the previous cytopenias, preventing additional cycles.
However, the patient’s overall condition gradually worsened. She developed hepatic dysfunction with AST at 245 U/L and ALT at 189 U/L (both more than five times the upper limit of normal) in the context of known liver metastases. Over the following two weeks, progressive multi-organ failure occurred. The primary tumor continued to grow aggressively locally despite radiotherapy, leading to tracheal compression and increasing dyspnea. The patient died from sudden respiratory and cardiac arrest approximately six months after initial symptom onset and three months after histological diagnosis.
The profound pancytopenia after a single cycle of reduced dose chemotherapy was more severe and lasted longer than usually expected, possibly due to the cumulative effects of previous immunosuppression, underlying SLE-related bone marrow issues, or hidden bone marrow involvement by MCC. The quick local tumor growth despite radiotherapy was also striking.
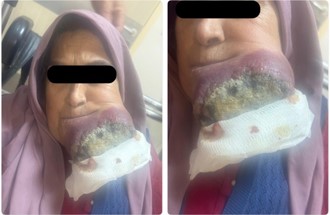
Figure 1: Rapidly enlarging mass with necrotic areas on the left jaw, demonstrating overlying skin erythema and focal ulceration characteristic of advanced Merkel cell carcinoma.
This case demonstrates the development of MCC in a patient who underwent long-term immunosuppressive therapy for SLE and APS. Although patients with systemic autoimmune diseases generally have an increased risk of malignancy, the evidence strongly indicates that immunosuppressive therapy, rather than the autoimmune disease itself, is the primary factor contributing to MCC development [3,5,6]. The biological plausibility is strong: MCC is mainly driven by MCPyV, whose oncoproteins are highly immunogenic foreign antigens that should be eliminated by a healthy T-cell response. Pharmacologic immunosuppression weakens this immune surveillance, enabling tumor cells expressing viral antigens to avoid destruction [2,4]. This explains why the highest MCC risk is seen in solid organ transplant recipients (23.8-fold), who receive the most intensive immunosuppression [3].
Our patient received multiple immunosuppressive agents over approximately 10 years, including azathioprine, rituximab, and prolonged corticosteroid exposure. Rituximab, a B-cell depleting agent, and azathioprine, an antimetabolite, both impair immune surveillance mechanisms critical for controlling MCPyV-driven tumorigenesis. This cumulative immunosuppressive burden likely represents the primary risk factor for MCC development in our patient, rather than SLE or APS per se.
A review of the medical literature reveals only two previously published cases of MCC in SLE patients. Nemoto et al. reported a 73-year-old Japanese woman with SLE and Sjögren’s syndrome on oral prednisolone plus methotrexate for 10 years who developed MCC with subsequent leukemic dissemination and complete bone marrow replacement by tumor cells [7]. Lentz et al. described a 55-year-old man with SLE who developed MCC with diffuse bone marrow infiltration causing pancytopenia [8]. Both cases involved patients receiving chronic immunosuppression, both developed bone marrow metastases, and both had fatal outcomes. Our case represents the third documented occurrence of MCC in an SLE patient and shares the common features of prolonged immunosuppressive therapy and an aggressive disease course. No cases of MCC in antiphospholipid syndrome patients have been reported.
The pancytopenia observed following chemotherapy presented significant diagnostic uncertainty. Multiple etiologies were plausible: chemotherapy-induced myelosuppression is expected with platinum/etoposide regimens; bone marrow metastasis is documented in MCC, particularly in immunosuppressed patients; and SLE-related autoimmune cytopenias were part of this patient’s history. The empirical response to high-dose corticosteroids and IVIG with platelet improvement might suggest an immune-mediated component. However, this response does not exclude bone marrow involvement, as the two previously reported MCC-SLE cases both developed bone marrow metastases [7,8].
Checkpoint inhibitor immunotherapy has become the standard first-line treatment for advanced MCC, with response rates over 50% [9]. However, immune checkpoint inhibitors can activate or worsen autoimmune conditions, posing a significant challenge for patients with pre-existing autoimmune diseases like SLE. In our patient, immunotherapy was postponed due to concerns about triggering a severe lupus flare. This situation highlights the therapeutic dilemma when managing MCC in patients with autoimmune diseases who need ongoing immunosuppression.
This case report offers several contributions to the literature. First, it is only the third documented instance of MCC in an SLE patient, adding to the very limited evidence available. Second, it provides detailed documentation of the 10-year immunosuppressive treatment history prior to MCC development, supporting the idea that cumulative immunosuppression, rather than autoimmune disease alone, increases MCC risk. Third, the case highlights the practical challenges of managing MCC in patients with complex autoimmune conditions, including contraindications to checkpoint inhibitors and the difficulty in diagnosing cytopenias. Fourth, the multidisciplinary approach involving rheumatology and oncology services sets a model for managing such complex cases.
This case report has several limitations. First, the lack of a bone marrow biopsy prevents definitive determination of the cause of pancytopenia, and bone marrow involvement by MCC cannot be ruled out. Second, MCPyV testing was not performed on the tumor specimen, which limits our ability to confirm a viral-driven pathogenesis. Third, since this is a single case report, we cannot establish causality between immunosuppressive therapy and MCC development, only a correlation that aligns with existing literature. Fourth, the absence of an autopsy restricts our understanding of the full extent of the disease at the time of death.
We present the third documented case of Merkel cell carcinoma in a patient with systemic lupus erythematosus, occurring in the context of chronic immunosuppressive therapy for both SLE and antiphospholipid syndrome. The main lessons from this case are threefold: first, long-term immunosuppression is likely the primary risk factor for MCC development in such patients, highlighting the importance of vigilant skin monitoring in all individuals on chronic immunosuppressive therapy; second, attributing cytopenias in complex immunosuppressed oncology patients requires careful consideration of multiple causes, including chemotherapy effects, bone marrow metastasis, and autoimmune disease flares; and third, managing MCC in patients with autoimmune diseases presents unique therapeutic challenges, especially because of the contraindication to checkpoint inhibitor immunotherapy.
Informed consent: Written informed consent was obtained from the patient before her death for the scientific use of clinical data and images of the lesion in the head and neck region. After her death, written consent was also obtained from her next of kin for publication of this case report.
Ethical statement Ethical committee approval was not required for this single case report per institutional guidelines.
Conflict of interest The authors declare no conflicts of interest.