Article Type: Case Report, Volume 2 Issue 2
*Corresponding author: Hamid Galehdari
Department of Biology, Faculty of Sciences, Shahid Chamran University of Ahvaz, Ahvaz, Iran.
Email: galehdari187@yahoo.com
Received: Nov 19, 2025 Accepted: Dec 15, 2025 Published: Dec 22, 2025
Citation: Saadati N, Zamani M, Galehdari H. Molecular and clinical characterization of Meckel-Gruber syndrome: A case study. Ann Case Rep Med Images. 2025; 2(2): 1057.
Copyright: Galehdari H et al. © All rights are reserved
Background: We aimed to report a rare clinical case of Meckel syndrome diagnosed by early ultrasound for prenatal diagnosis with characteristic signs, and molecular genetic validation using whole exome sequencing (WES) to make an update on genetic heterogeneity and polymorphism of this entity in our population.
Case presentation: A young woman, 31 years old in a consanguineous marriage, in her first pregnancy had to terminate the pregnancy at 20 weeks because she had been diagnosed prenatally during routine anomaly scan with encephalocele and polydactyly in fetus, which are characteristic and overlapping symptoms of Meckel-Gruber syndrome, Joubert syndrome, and Bardet-Biedel syndrome. She was referred for genetic counseling and subsequent molecular diagnostics of aborted fetus.
Conclusion: We found a novel homozygous alteration in the MKS1 gene with the reference number NM_01777.4, derived from fetal muscle tissue that was validated by Sanger sequencing of the parents, both of whom were heterozygous. The coordination of the variant is chr.17:5629166 (G>A), c.598 C>T / p.Pro200Ser. This alteration is classified as likely pathogenic according to ACMG, which is supported by other mutation prediction programs. This is the first clinical and molecular manifestation of Meckel syndrome in southwestern Iran, which was initially diagnosed prenatally based on morphological ultrasound examination and subsequently subjected to molecular genetic analysis using Whole Exome Sequencing (WES).
Keywords: Meckel-Gruber-syndrome; MKS1 gene; Iran; Whole exome sequencing.
The worldwide incidence of Meckel-Gruber syndrome (MGS) is 1 in 140,000 live births, except in Finland, where the incidence is 1:50. However, it is generally considered a rare fatal disorder with an autosomal recessive inheritance pattern [1-4].
Our understanding of the pathogenesis of MGS has progressed over the last few years since the identification of the first gene mutation in the MKS1 gene, which codes for a ciliary protein. Nine additional genes have subsequently been identified, all coding for the same kind of ciliary protein: TMEM216 (MKS2), TMEM67 (MKS3), CEP290 (MKS4), RPGRIP1L (MKS5), CC2D2A (MKS6), NPHP3 (MKS7), TCTN2 (MKS8), B9D1 (MKS9), and B9D2 (MKS10), which demonstrates the heterogenic nature of MGS caused by cilia and flagella dysfunction [5-12]. Impaired ciliary biology is however not specific [13]. A huge group of disorders that are commonly referred to as ciliary defects share this basic feature. Polycystic liver and kidney disease, Bardet-Biedl syndrome, Alström syndrome, and Joubert syndrome are a few more to be included in the list. Causally, pathogenic alterations in the MKS gene family also lead to other diseases, i.e., MKS1 in Bardet-Biedl, TMEM216 in Joubert syndrome, TMEM67 in Joubert syndrome and nephropathy, CEP290 in non-syndromic retinal dystrophy, salivary-Lukken syndrome, nephronephritis, Joubert syndrome and Bardet-Biedl syndrome, RPGRIP1L and Joubert syndrome, and CC2D2A in Joubert syndrome [14-21].
The determinants of the final clinical phenotype are not yet fully understood, although there is growing evidence that the diseases exhibit a spectrum of clinical severity, in part determined by the severity of the ciliary defect. MKS is the most severe form of the clinical spectrum, whereas kind of mutation in the same gene is crucial for the outcomes and phenotypes though with some overlap [22].
The genetic heterogeneity of FMD is still not fully understood. In fact, a recent publication showed that only half of the cases are due to mutations in one of the genes presently known to be involved [23].
A young woman, thirty-one years old and her husband’s cousin during her first pregnancy, had to terminate the pregnancy at 20 weeks due to a prenatal diagnosis of encephalocele and was referred for genetic counseling. External examination revealed encephalocele, hypertelorism, low-set ears, polydactyly and syndactyly of the extremities (Figure 1).
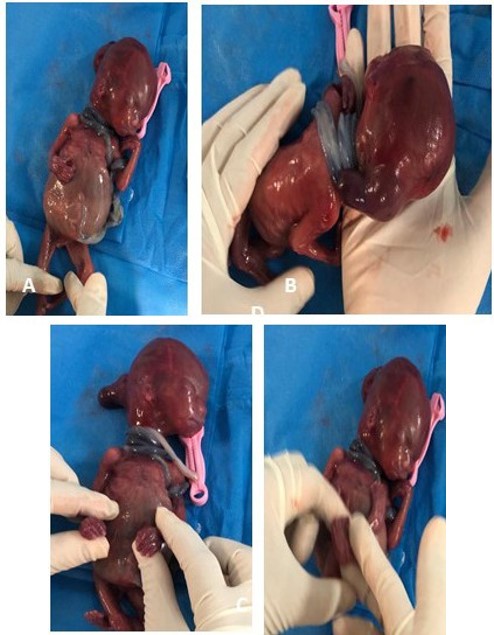
Figure 1: The aborted fetus showed clubfoot and abdominal distension (A), encephalocele (B), syndactyly of the hands (C), and polydactyly of the feet.
Using WES we identified a novel homozygous variant in the MKS1 gene, which altered the B9 domain of the gene product (p.Pro200Ser). This change is according different mutation prediction program classified as likely pathogenic. Regarding AR pattern of MGS, parents have to be obligate carrier (heterozygous), what it was confirmed by Sanger Sequencing of parents (Figure 2).
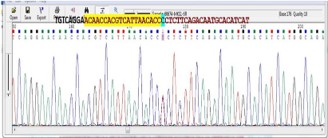
Figure 2: Sanger sequencing of parents reveals their heterozygous status for disease causing mutation.
Prenatal ultrasound is the most easily accessible technique for the diagnosis of Meckel-Gruber syndrome and is available in two (2D), three (3D), and four-dimensional (4D) formats. The 4D however is useful for assessing different and subtle abnormalities [24]. MRI is a valuable adjunct to ultrasound for the assessment of fetal anomalies in cases of severe oligohydramnios. It is generally reserved when ultrasound is inconclusive or insufficient for management decisions [25,26]. However, similarity of MGS with other ciliopathies requires an exact determination of the mutated gene [27].
We present here molecular diagnosis of MGS according to prenatal signs of the fetus. The p.Pro200Ser variant in the MKS1 gene is located in the B9 domain, which is enriched with disrupting changes and well reported in MKS1-related ciliopathies [28]. Previous study showed that the domain proteins Mks1, B9d1, and B9d2 physically interact. Knowingly, the p.Ser101Arg mutation abolishes the ability of B9d2 to interact with Mks1, suggesting that alterations in this domain disrupt B9d2 function. Further evidence showed that B9d1 is required for intact Hedgehog (Hh) signaling, ciliogenesis, and the localization of ciliary proteins, which are essential components of a B9 protein complex whose disruption causes Meckel syndrome [29].
Comparative structure analysis of P200S variant via SWISSMODEL® showed the impact of this change on the N-terminal of MKS1 (Figure 3).
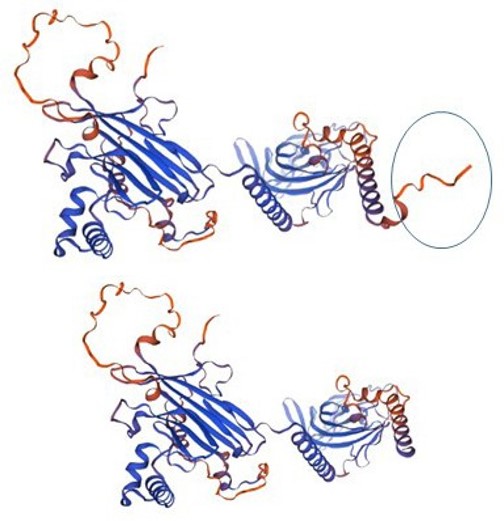
Figure 3: The left structure shows the normal MKS1 with N-terminus (circled) and the right structure after the P200S modification.
Whole Exome sequencing is a widely used Next-Generation Sequencing (NGS) method that sequences the protein-coding fraction of the genome for finding mutation causing disease in such complex and similar to many other syndromes. The human exome consists of fewer than 2% of the genome but harbors 85% of disease-associated variants known to date and is therefore an economical alternative to whole-genome sequencing [30].
This is the first report of Meckel Gruber syndrome in Iran, which is characterized by genetic heterogeneity. The finding was firstly supported through ultrasound and clinical observation after abortion as critical diagnostic tool, and verified using WES and Sanger sequencing, subsequently. Overall, based on the concordance of clinical and molecular findings, we believe that the identified variant may be pathogenic, which, however, requires further functional analyses.
Acknowledgments: Hereby, we extend our gratitude to all the colleagues who assisted us in this research project.
Conflicts of interest: The authors declare that there is no conflict of interest.