Article Type: Case Report, Volume 2 Issue 2
*Corresponding author: Ha Vu Thi
Department of Medical Biology and Genetics, Hanoi Medical University, No1, Ton That Tung, Dong Da, Hanoi, Vietnam.
Email: vuthiha@hmu.edu.vn
Received: Sep 25, 2025 Accepted: Oct 23, 2025 Published: Oct 30, 2025
Citation: Mai TTM, Nguyen HC, Vu AH, Nguyen TC, Ha Vu T. Muscle weakness associated with CVA1 gene mutation: Clinical insights from a vietnamese case. Ann Case Rep Med Images. 2025; 2(2): 1045.
Copyright: Ha Vu T et al. © All rights are reserved
Congenital generalized lipodystrophy with CAV1 variants is a group of conditions characterized by abnormal adipose tissue distribution, including complete or partial loss of adipose tissue or accumulation in internal organs. In males, proximal muscle weakness is commonly associated with Duchenne muscular dystrophy or limb-girdle muscular dystrophy. Here, we present a case of proximal muscle weakness linked to a mutation in the CAV1 gene. A 34-yearold male patient exhibited symptoms consistent with Duchenne/Trendelenburg syndrome, characterized by irregular coordination of the shoulders and pelvic bones, resulting in a labored gait and challenges in hip, shoulder, and hand joint movements. Clinical Exome Sequencing, along with subsequent Sanger sequencing, was performed and revealed that the patient had a heterozygous in-frame deletion in the CAV1 gene (NM_001753.5: c.87_89del) (NP_001744.2:p.Asn29del). This variant had not been previously reported. Additionally, his 3-year-old daughter, who suffered from a similar disorder when he was a small child, had a similar genotype. This is the first reported case of autosomal dominant CAV1-associated muscle weakness combined with congenital generalized lipodystrophy in Vietnam.
Keywords: Total/partial body lipodystrophy; CAV1 gene mutation; Duchenne/Trendelenburg signs; Muscle weakness.
CAV1 (Caveolin 1) is a protein-coding gene responsible for encoding caveolin-1 protein, which was found within the caveolae microdomains of the plasma membrane and plays crucial roles in various signaling pathways. Furthermore, caveolin-1 is present in lipid droplets of adipocytes. Pathogenic variants in CAV1, whether heterozygous or homozygous, are related to rare and diverse disorders such as pulmonary arterial hypertension, neonatal progeroid syndrome, and congenital generalized lipodystrophy [1].
Congenital Generalized Lipodystrophy (CGL), also known as Berardinelli-Seip syndrome, is a rare condition characterized by virtually absent fatty tissue. Fat loss is usually evident either at birth or within the first year of life, although some patients may be diagnosed later. Patients exhibit a lack of subcutaneous fat but may have enlarged livers and protruding abdomens in infancy. It is typically inherited in an autosomal recessive manner, depending on the causative gene: AGPAT2, BSCL2, CAV1, or PTRF [2-4]. Congenital generalized lipodystrophy related to the CAV1 gene may present with short stature, vitamin D resistance, hypocalcemia, and magnesium deficiency [5]. A heterozygous frameshift mutation of the CAV1 gene associated with atypical partial lipodystrophy and hypertriglyceridemia has been reported [6,7]. Another study found a heterozygous CAV1 mutation identified by exome sequencing associated with a novel neonatal-onset lipodystrophy syndrome [8]. In this study, we report a rare case of a heterozygous in-frame deletion of the CAV1 gene associated with muscle weakness and congenital generalized lipodystrophy, with detailed clinical characterization, genetic analysis, and autosomal dominant inheritance.
A 34-year-old male patient has been experiencing a limp gait, difficulty walking upstairs, or standing up at 3-5 years. These symptoms have been progressively worsening. There was no intellectual disability. He came to our attention with a petite stature, measuring 141.2 cm in height and weighing 34 kg, with a body mass index of 17.6. His gait is cumbersome, indicative of Duchenne/Trendelenburg syndrome (Figure 1), with deformed elbows, flexed wrists, and clenched fists when extended. His overall bone density was severely low, with a T-score of -5.4, and he had a vitamin D deficiency of 17.1 ng/ml. His blood test showed a mild increase in triglycerides (2.78 mmol/L), total cholesterol (6.34 mmol/L), and LDL-C (4.04 mmol/L). Abdominal ultrasonography revealed hepatic steatosis but otherwise normal viscera. Whole-body fat mass assessed by the DEXA method shows no subcutaneous or visceral fat. X-ray images of the patient show slender humerus bones and deformed elbow joints, with normal hip joint and ribs cartilage calcification (Table 1).
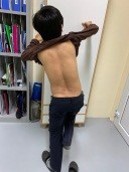
Figure 1: The Duchenne/Trendelenburg sign of the patient.
| Clinical characteristics and testing results | Result | Reference range |
|---|---|---|
| Body mass index (kg/m2) | Underweight (BMI=17.6) | 18.5-24.9 |
| Lipodystrophy onset | Early infancy | |
| Squat | Failed to perform | |
| Proximal muscle weakness | Positive tabouret sign | |
| The Duchenne de Boulogne sign | Positive sign | |
| The Trendelenburg sign | Positive sign | |
| The Duchenne/Trendelenburg sign | (+) | |
| Flexor tendon shortening | (+) | |
| X-ray: Slender humerus bones and elbow joints deformity | (+) | |
| Osteoporosis (DEXA scan test) bone mineral density at lumbar spine T-score | T-score: - 5.4 | >-1 |
| No subcutaneous or visceral fat (DEXA) | 0.0 | |
| Glucose | 5.02 mmol/L | 4.1-5.9 mmol/L |
| Cholesterol | 6.34 mmol/L | <5..2 mmol/L |
| Triglyceride | 2.78 mmol/L | <1.7 mmol/L |
| High density lipoprotein cholesterol | 1.04 mml/L | >0.9 mml/L |
| Low density lipoprotein cholesterol | 4.04 mml/L | <3.4 mml/L |
| 25(OH)Vitamin D (D3) | 17.6 ng/mL | >30 ng/mL |
| Creatinine kinase | 84.9 U/L | 30-170 U/L |
| Total calcium | 2.40 mmol/L | 2.1-2.6 mmol/L |
| Ionized calcium | 1.20 mmol/L | 1.17-1.29 mmol/L |
His father also had a petite stature and showed signs of muscle weakness; he passed away without a clear diagnosis. His 3-year-old daughter also exhibits muscle weakness similar to his own during childhood. Other family members, such as his mother, sister, and wife, are all normal.
A peripheral blood sample was taken from the patient for Clinical Exome Sequencing. The result of the genetic analysis showed that the patient had a heterozygous in-frame deletion of the CAV1 gene (NM_001753.5: c.87_89del) (NP_001744.2:p. Asn29del). This variant was searched using in silico tools and public domain libraries (ClinVar, gnomAD, HGMD) to confirm whether it had been previously reported. Based on this process, we confirmed that this variant had not been previously reported in any literature or clinical variant databases. Sanger sequencing revealed that his 3-year-old daughter, who suffers from the same disorder as the patient did during his childhood, had a similar genotype. Unfortunately, we don’t have the genotype of his father, who had similar symptoms. Other members of his family, including his mother, sister, and wife, have normal genotypes according to Sanger sequencing results (Figure 2) and normal phenotypes.
Family pedigree showing the dominant inheritance of the CAV1 variant in the proband (II.3, indicated by arrow) and his daughter (III.3), both diagnosed with CAV1. His father who has died with symptoms of muscular but non-diagnosis. His mother (I.2), his wife (II.4), his older sister (II.2) are also shown no mutation.
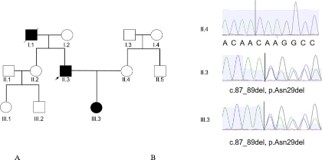
Figure 2: The patient’s family pedigree (A) and Sanger’s sequencing result (B).
Differential diagnosis
The patient exhibits many symptoms similar to Duchenne muscular dystrophy, a hereditary muscle disorder described by Guillaume Duchenne in 1858. These symptoms include a male sex, Duchenne/Trendelenburg gait, early disease onset, proximal muscle weakness, positive Tabouret sign, and flexor tendon shortening. However, there are some inconsistent features with Duchenne muscular dystrophy, such as normal creatine kinase levels and nondetected mutation on the DMD gene by DMD gene sequencing.
The goal of treatment was decreasing the muscle weakness progression of muscular dystrophy. The first-line treatment involves steroids (deflazacort or prednisone) combined with kinesitherapie and ergotherapie, aimed at maintaining joint flexibility. In cases of muscular dystrophy with spinal deformities, the orthopedic doctor plays a significant role. In our patient, due to severe osteoporosis and vitamin D deficiency, we chose a bone resorption inhibitor (alendronate 70 mg weekly), supplemented with vitamin D and combined it with daily exercise to prevent joint stiffness and muscle atrophy from reduced mobility.
According to the literature, congenital generalized lipodystrophy manifests in four subtypes, with types 1 and 2 being more prevalent, and types 3 and 4 being rare. Specifically, type 3 is uncommon. Karhan AN et al. conducted a study on a consanguineous family from Turkey with type 3 lipodystrophy due to a CAV1 gene mutation encoding Caveolin-1. The patients ranged in age from 8 months to 18 years and exhibited insulin resistance, severe hypertriglyceridemia, and polycystic ovary syndrome. Osteoporosis and severe dysphagia causing swallowing difficulty were observed in two patients aged 15 and 18, without any cardiac manifestations [7]. A study by Lima JG in 2018 on bone density in patients with congenital generalized lipodystrophy type 2 found increased bone mass in the pelvis [9]. Our patient, along with those reported by Karhan AN, exhibited severe early-onset osteoporosis despite the phenotypic characteristics of type 3 lipodystrophy, including short stature, vitamin D deficiency, and osteoporosis, which are consistent with the reported clinical features.
In our study, the patient underwent a comprehensive medical history assessment, clinical examination, and evaluation of gait-related signs. He presented with a Duchenne/Trendelenburg gait and proximal muscle weakness. Combined with genetic analysis revealing a CAV1 gene mutation with a heterozygous variant on chromosome 7, the patient was diagnosed with partial lipodystrophy with a heterozygous variant CAV1 type 3. He has a short stature, measuring 141.2 cm in height and weighing 34 kg, with a body mass index of 17.6, resembling the phenotype of different types of lipodystrophy as classified and described by author Iram Hussain in 2008 [10]. Disorder of stability in the vertical plane of the pelvis is observed, attributed to muscle weakness. The patient exhibits evident muscle weakness in assessment tests; he was unable to squat or stand up independently. Additionally, while walking, he displays a limp in the shoulders and pelvis. These signs indicate a combined shoulder and pelvic limp, resulting in a Duchenne/Trendelenburg gait.
The signs related to muscles include weakness in the proximal muscles (shoulder and hip joints), difficulty walking, inability to squat, and the inability to stand up independently, all suggestive of muscle-tendon damage. Additionally, Achilles tendon stiffness contributes to difficulty in walking. There is also difficulty in lifting the shoulders and maintaining balance, with challenges encountered in removing tight-fitting clothing. The presence of Trendelenburg sign while walking and difficulty standing up from a squatting position further suggests proximal muscle weakness. This musculoskeletal condition observed in our patient has not been described in the literature.
Congenital generalized lipodystrophy constitutes a rare group of disorders with diverse etiologies. It is typified by abnormal fat tissue distribution, involving either peripheral and subcutaneous fat loss or visceral fat accumulation. In a departure from typical presentations, our patient showed no subcutaneous or visceral fat based on DEXA imaging. Muscle weakness has been documented in various studies. For example, in patients with myotonic dystrophy type 1, muscle weakness has been linked to the extent of alternative splicing of exon 29 of the CAV1 gene [11]. Furthermore, a study reported two siblings diagnosed with congenital generalized lipodystrophy and congenital muscular weakness without CAV1 mutations or other common genes. Our patient displayed typical lipodystrophy characteristics of type 3, including short stature, vitamin D deficiency, and severe osteoporosis, aligning with documented clinical profiles. However, our patient also exhibited a Duchenne/ Trendelenburg gait, indicating significant movement difficulty and an unsteady gait.
This is first case of congenital generalized lipodystrophy with CAV1 gene mutation observed in Vietnam, significantly affecting the patient’s motor function. The patient presents with a Duchenne/Trendelenburg gait, dysfunction of hand movement, a knee joint disorder, severe osteoporosis, and vitamin D deficiency. The diagnosis is established through genetic analysis conducted on both the patient and his daughter. In the future, early research on congenital generalized lipodystrophy is imperative to mitigate serious complications such as proximal muscle weakness, severe osteoporosis, and joint deformities, with the aim of minimizing motor dysfunction.
Author contributions: Tam Thi Minh Mai, Ha Thi Vu coordinated the study. Huu Cong Nguyen, Anh Hong Vu, Trung Cong Nguyên involved in clinical diagnosis and collected patient data. Tam Thi Minh Mai, Ha Thi Vu performed genetic analysis. Tam Thi Minh Mai, Ha Thi Vu interpreted the results and wrote the manuscript. All authors have read the manuscript and approved of the final version for publication.
Conflict of interest: The authors declare no conflict of interest.
Data availability statement: The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowlegments: The authors sincerely thank the valuable contributions of the patients and his family.